|
|
|
|||||
|
||||||
| 治療的大門已開啟-認識遺傳性視網膜疾病 |
|
|
|||
| 眼科部 劉沛綱 助理教授 (112年4月) | |||
|
32歲的陳小姐於七年前開始感覺夜間駕駛交通工具很吃力,其父親被診斷患有”夜盲症”,因此懷疑自己遺傳到父親的夜盲症,更擔心未來自己的小孩會不會也被遺傳到相同疾病,因此偕同未婚夫來到本院眼科檢查。經散瞳詳細眼底檢查(圖一)後證實,陳小姐患有視網膜色素變性(retinitis pigmentosa)。基因檢測發現陳小姐帶有顯性遺傳之基因突變,因此她所生的每一個孩子都有50%的機率會患病。
遺傳性視網膜失養症(inherited retinal
dystrophy)可以透過家族遺傳或隨機突變引起,並以不同的遺傳形式傳給後代。視網膜色素變性則是最常見之遺傳性視網膜失養症,在台灣盛行率約為千分之一,大多數患者雙眼皆會受影響且持續惡化,最終可能導致失明。症狀包含夜盲、視覺障礙、對物體明暗對比或顏色分辨能力下降、管狀視野狹窄。眼底表現為在視網膜上會出現眾多骨針狀色素斑點,視網膜動脈縮小、視神經萎縮等變化。有些患者會合併聽力障礙而導致視力聽力皆受影響。 萊伯氏先天性黑朦症為一種早發性的遺傳性視網膜失養症,臨床表現為早發且嚴重的視覺功能障礙,病程大多開始於嬰幼兒期,臨床表現是出生不久後即有嚴重之視力下降,眼球震顫,以及眼手徵兆(包括戳眼、壓眼以及揉眼睛三個特徵)。過去此疾病並無藥物可治療。在次世代基因定序技術(NGS)的幫助之下,臨床醫師評估確認臨床診斷後三個月內即可找出致病基因及突變位點。 2017年美國FDA核准第一個眼科基因治療藥物(Luxturna),此藥物以無法複製之病毒(AAV2)做為載體,攜帶RPE65蛋白的正確基因序列(cDNA)。將藥物顯微注射到視網膜下使眼球的細胞可以合成具正常功能之蛋白質,從而恢復視覺功能。此藥物可用於治療因RPE65基因突變引起之萊伯氏先天性黑朦症或視網膜色素變性,食藥署於2023年1月已核准此藥物,本院眼科部提供此基因藥物之治療。除Luxturna外,目前亦有多項眼科基因治療藥物在美國已經進展至第二、三期臨床試驗,包含斯特格氏病,尤塞氏綜合症,脈絡膜失養症等。 遺傳性視網膜疾病的診斷需要完整的視覺電生理檢查、影像學檢查等。於精準醫學的新時代,此類疾病之治療目標包含找出致病基因,了解病程及預後,進而早期發現和治療以延緩視覺功能惡化達到保存視力之目的。本院眼科部提供基因檢測、基因報告判讀及解說、以及基因藥物治療等服務。基因檢測除了找出致病基因進而接受基因治療外,亦可預判後代患病機率進而達到優生學之目的。建議具有上述症狀及有家族史之病友至眼科門診接受詳細的檢查及評估。
※本文章同意非營利行為之轉載,轉載時不得編輯、調整、增刪、加註、改寫或以其他方式變更本文章內容,並註明供稿來源。
|
|||
|

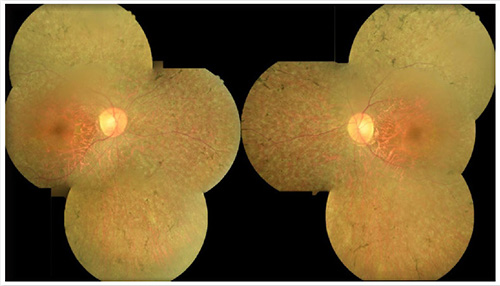 (圖一)
(圖一)| 高雄市三民區自由一路100號(地圖) │ 聯絡信箱│網頁維護:kmuj@kmuh.org.tw 高醫醫訊雜誌社 版權所有 © 2013 KMUH All Rights Reserveda 建議使用IE 9.0以上1024×768為最佳瀏覽 |